Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska tundra ecosystem
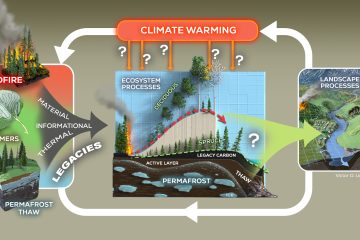
How soil microbial communities contrast with respect to taxonomic and functional composition within and between ecosystems remains an unresolved question that is central to predicting how global anthropogenic change will affect soil functioning and services. In particular, it remains unclear how small-scale observations of soil communities based on the typical volume sampled (1-2 grams) are generalizable to ecosystem- scale responses and processes. This is especially relevant for remote, northern latitude soils, which are challenging to sample and are also thought to be more vulnerable to climate change compared to temperate soils. Here, we employed well replicated shotgun metagenome and 16S rRNA gene amplicon sequencing to characterize community composition and metabolic potential in Alaskan tundra soils, combining our own datasets with those publically available from distant tundra and temperate grassland and agriculture habitats. We found that the abundance of many taxa and metabolic functions differed substantially between tundra soil metagenomes relative to those from temperate soils, and that a high degree of OTU-sharing exists between tundra locations. Tundra soils were an order of magnitude less complex than their temperate counterparts, allowing for near- complete coverage of microbial community richness (~92% breadth) by sequencing, and the recovery of twenty-seven high-quality, almost complete (>80% completeness) population bins. These population bins, collectively, made up to ~10% of the metagenomic datasets, and represented diverse taxonomic groups and metabolic lifestyles tuned toward sulfur cycling, hydrogen metabolism, methanotrophy, and organic matter oxidation. Several population bins, including members of Acidobacteria, Actinobacteria, and Proteobacteria, were also present in geographically distant (~100-530 km apart) tundra habitats (full genome representation and up to 99.6% genome-derived average nucleotide identity). Collectively, our results revealed that Alaska tundra microbial communities are less diverse and more homogenous across spa tial scales than previously anticipated, and provided DNA sequencesof abundant populations and genes that would be relevant for future studies of the effects of environmental change on tundra ecosystems.